Overview of the HLA3D
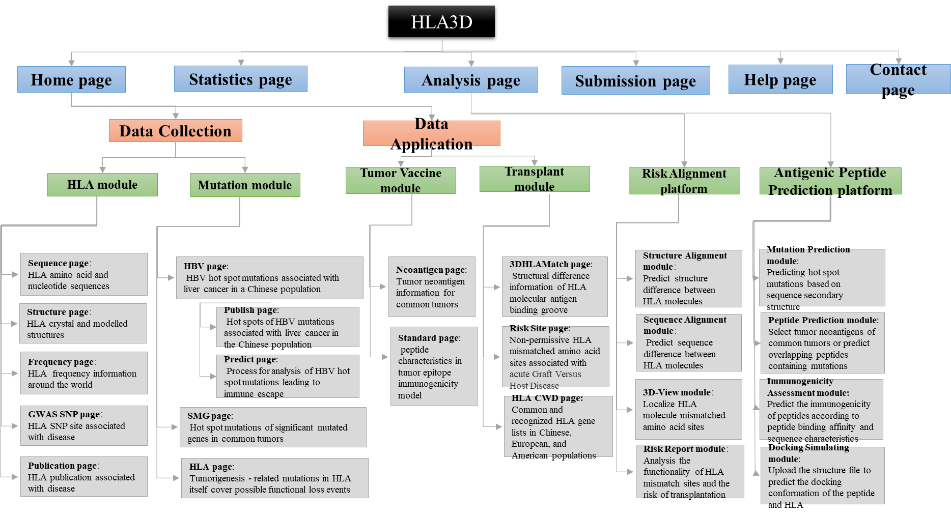
Figure1. The architecture of HLA3D. HLA3D toolkit includes twenty interfaces and two useful pipelines.
| Data Type | Data Sources | Description | External Links | |
|---|---|---|---|---|
| 1 | Sequence | IMGT/HLA | The IPD-IMGT/HLA Database provides a specialist database for sequences of the human major histocompatibility complex (MHC) and includes the official sequences named by the WHO Nomenclature Committee For Factors of the HLA System.[1] | https://www.ebi.ac.uk/ipd/imgt/hla/intro.html |
| 2 | Structure | PDB | As a member of the wwPDB, the RCSB PDB curates and annotates PDB data.[2] | https://www.rcsb.org/ |
| 3 | Frequency | ANFD | The Allele Frequency Net Database (AFND) provides the scientific community with a freely available repository for the storage of immune gene frequencies in different worldwide populations.[3] | http://www.allelefrequencies.net/default.asp |
| 4 | GWAS SNP | CAUSALdb | CAUSALdb integrates large numbers of GWAS summary statistics and identifies credible sets of causal variants by uniformly processed fine-mapping.[4] | http://mulinlab.tmu.edu.cn/causaldb/index.html |
| 5 | Publication | PubMed | PubMed® comprises more than 30 million citations for biomedical literature from MEDLINE, life science journals, and online books. Citations may include links to full-text content from PubMed Central and publisher web sites. | https://pubmed.ncbi.nlm.nih.gov/ |
| 6 | 3DHLAMacth | Manual collection | We predicted the structural differences in the antigen-binding slots among all HLA constructs in the HLA3D Toolkit to help investigators determine the optimal donor. | |
| 7 | Risk Sites | Manual collection | We collected amino acid mismatch sites reported in the literature that are associated with higher aGVHD risk, and annotated HLA amino acid sites that bind to antigenic peptides and TCR. | |
| 8 | HLA CWD Catalog | Manual collection | We collected ASHI, EFI, Chinese CWD Catalog to provide information on HLA Frequecy status in American, European and Chinese populations. | |
| 9 | Neoantigen | Manual collection | We collected hot spot mutations in common tumors and used NetMHCpan4.0 to predict the binding affinity of mutant peptides to all common HLA molecules in the HLA3D Toolkit. | |
| 10 | HLA Mutation | Manual collection | We collected HLA mutations detected in common tumors to help researchers understand the mechanisms of tumorigenesis.[5] | |
| 11 | HBV Mutation | Manual collection | We collect HBV mutations reported in the literature that are associated with liver cancer in Asian population to promote immunotherapy for HBV. | |
| 12 | SMG Mutation | Manual collection | We collected Hotspot mutations of Significant mutated gene detected in common tumors to help researchers predict antigenic peptides.[6] |
| Tool | Description | |
|---|---|---|
| 1 | 3Dmol[7] | Molecular visualization |
| 2 | PSIPRED 4.0[8] | Predict Secondary Structure |
| 3 | ClustalW2[9] | Protein multiple sequence alignment |
| 4 | PeptideBuilder[10] | Construction of peptide conformation |
| 5 | CodockPP[11-13] | A multistage protein-protein docking program based on shape complementarity, knowledge-based scoring function and site constraint. |
| 6 | MHCflurry[14-15] | MHCflurry is an open source package for peptide/MHC I binding affinity prediction. |
Data Feature
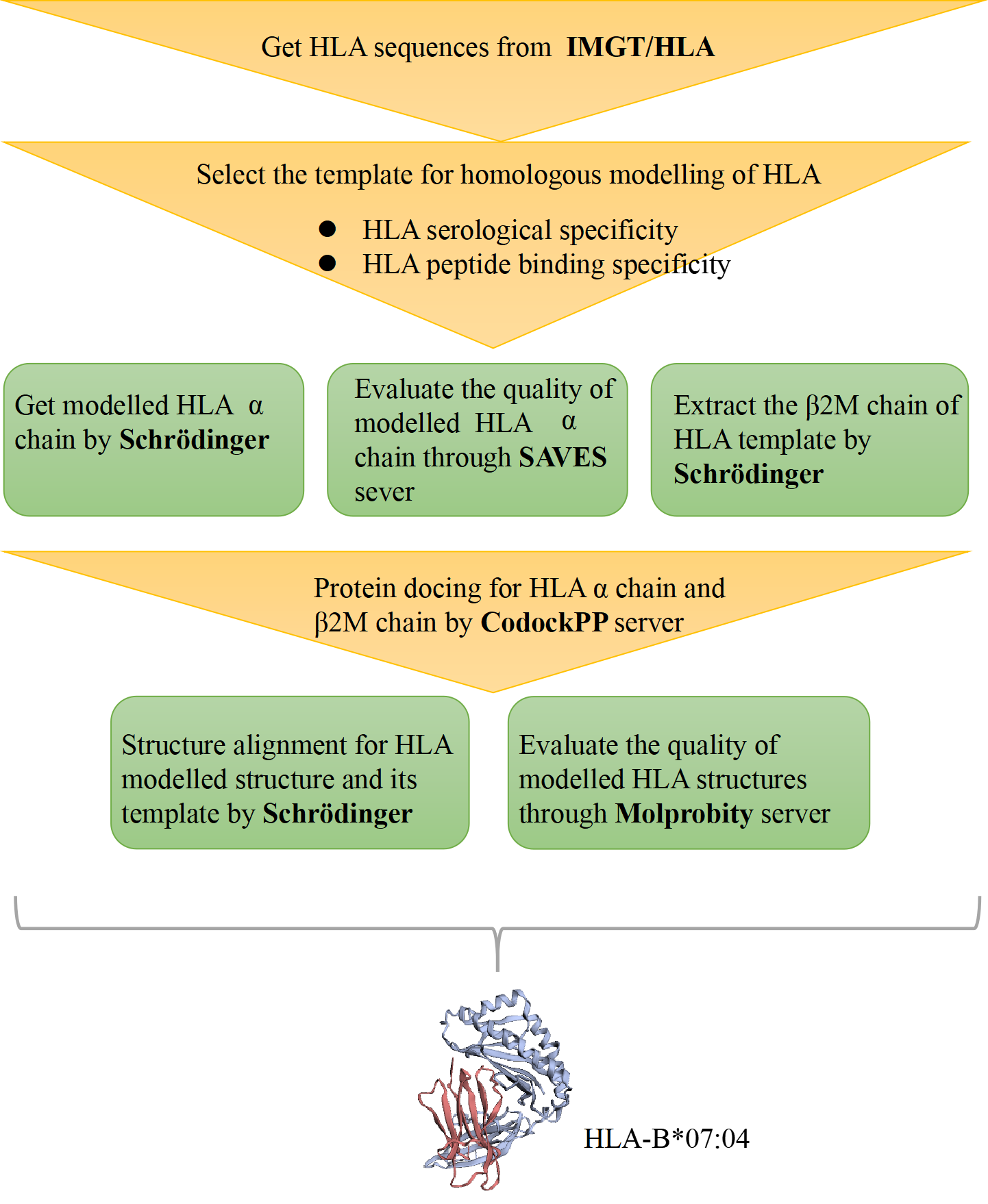
Figure 1. The process of HLA structure construction.
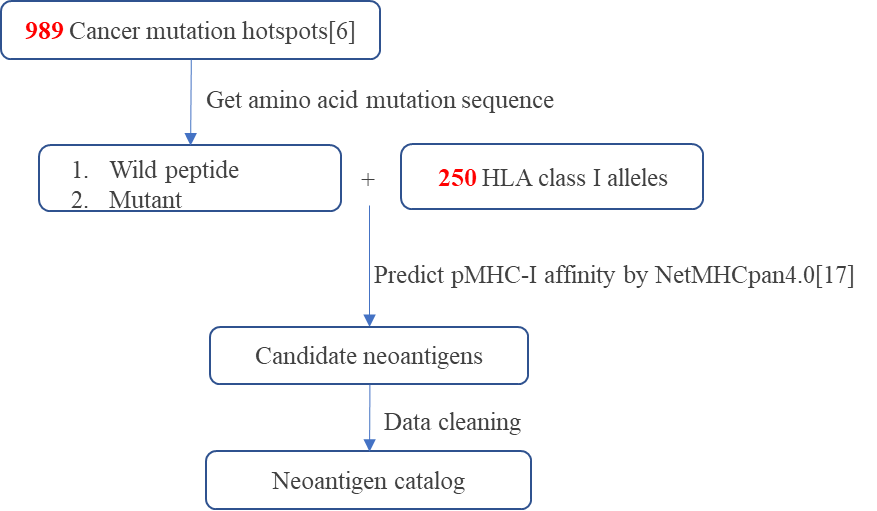
Figure 1. The process of predicting high affinity neoantigens for common tumors.
Step-1 Enter Query information
i.Gene
Mutated gene in tumor. The HLA3D Toolkit provides the prediction results of 127 significant mutated genes in 11 common tumors[6].
ii.Mutation
Mutation of a gene. The HLA3D Toolkit provides the prediction results of 989 hot spot mutations in 11 common tumors[6], all of which are missense mutations.
iii.HLA Allele
The HLA allele that binds the mutant peptide. Combined with the HLA alleles of the mutant peptide, the HLA3D Toolkit provides predictions of all common HLA class I alleles in the US, European and Chinese populations, a total of 250 alleles.
iv.Peptide
A peptide sequence containing a mutation. High-affinity peptides of 8-11 amino acids in length were predicted in HLA3D Toolkit, including wild-type and mutant types.
i.Cancer
The type of tumor that contains the mutant gene.
ii.Gene
The gene that contain the predicted mutation.
iii.Mutation
The mutation that produces an antigenic peptide.
iv.State
This indicates whether the mutant peptide is wild-type or mutant.
v.Peptide
Sequences of mutant peptides.
vi.Position
The location of the mutation.
vii.%Rank_EL
Rank of the predicted binding score compared to a set of random natural peptides. This measure is not affected by inherent bias of certain molecules towards higher or lower mean predicted affinities. Strong binders are defined as having %rank<0.5, and weak binders with %rank<2.[17]
viii.BindLevel
SB: Strong Binder, WB: Weak Binder.[17]
ix.HLA
The HLA allele used to predict affinity.
x.Validation
Users can click "Search" to jump to the IEDB database to check whether the mutated peptide has experimental data.
For example:
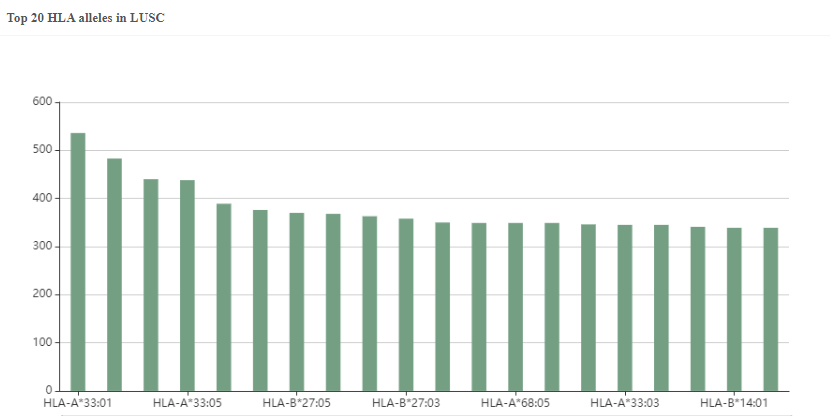
Figure 2. The predicted top20 HLA alleles with the most neoantigens in LUSC.
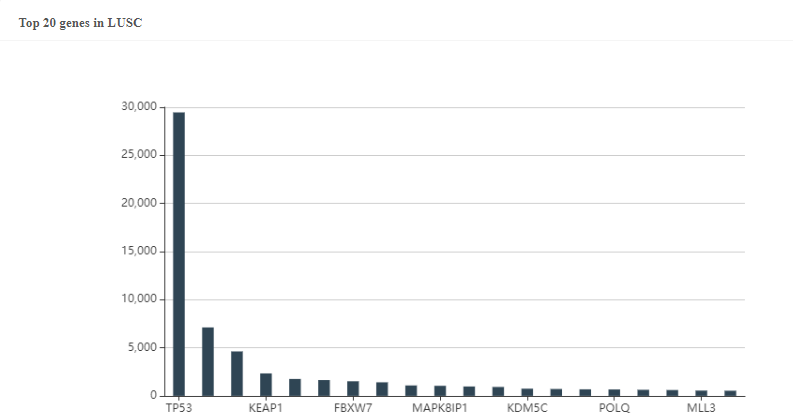
Figure 3. The predicted top20 genes with the most neoantigens in LUSC.
Step-1 Enter Query information
i.HLA Allele
HLA alleles for structural alignment.
ii.Aligned Allele
Another HLA allele for structural alignment.
iii.Gene Pair
You can query the structural difference between HLA-A*02:01 and HLA-A*11:01 by entering the form of gene pairs, such as "A* 02:01_A *01:01".
iv.Upload a file
You can upload a TXT file containing multiple gene pairs, one gene pair per line. You can click “Explanation” below the input box of the 3DHLAMacth page to get the “example.txt”.
i.Gene
HLA alleles for structural alignment.
ii.Aligned Gene
Another HLA allele for structural alignment.
iii.Structure
The HLA structure used for comparison in the HLA3D Toolkit. Some HLA genes have multiple PDB structures.
iv.Aligned Structure
The HLA structure used for comparison in the HLA3D Toolkit. Some HLA genes have multiple PDB structures.
v.RMSD Score
Used to indicate the structural difference of HLA antigen binding groove, the larger the value, the greater the difference.
Step-1 Enter Query information
i.Risk Position
The location of structural difference sites of HLA molecules.
ii.Alignment Position
The position of amino acid sequence difference sites of HLA molecules.
i.Alignment position
The position of amino acid sequence difference sites of HLA molecules.
ii.Risk position
The location of structural difference sites of HLA molecules.
iii.Location
This indicates the location of the HLA mismatch site on the secondary structure of the HLA molecular antigen binding groove.
iv.Pocket
This indicates the location of the mismatch site in the HLA antigen binding groove, which usually has six pockets of A-F.
v.Function
This indicates whether the HLA mismatch site interacts with the peptide or T cell receptor (TCR).
vi.Risk
"Risk" indicates that this position is an HLA mismatch site associated with acute graft-versus-host disease that has been reported in the literature.
vii.HLA Locus
The HLA locus of this mismatch site has been reported in the literature.
viii.PMID
PubMed ID of relevant literature.
Analysis
Introduction
Based on the knowledge of structural difference immunogenicity and sequence difference immunogenicity of HLA molecules, we designed Risk Alignment pipeline for users by integrating different resources and tools to help users quickly assess the transplantation risk of HLA unrelated mismatched donors.
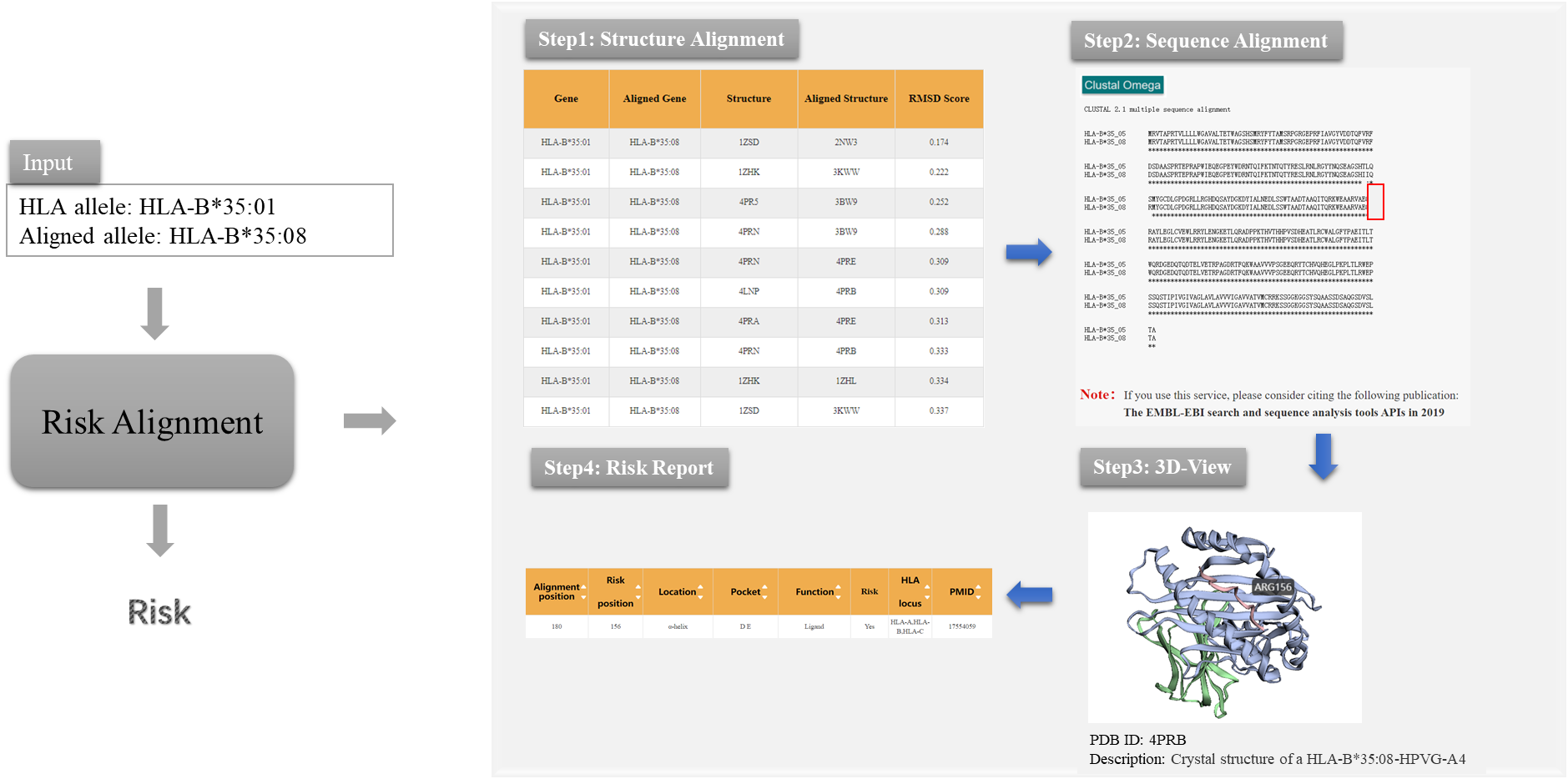
Figure 1. The prediction process of Risk Alignment pipeline in HLA3D
As the picture shows, Risk Alignment pipeline provides users with the following functions: (i) Structure Alignment This module aims to help users get the information on the conformational differences of HLA antigen binding groove. 3DHLAMatch page shows the conformational differences of antigen-binding pocket of all HLA structures in the HLA3D Toolkit, represented by RMSD value. The larger the RMSD, the greater the HLA molecular differences. Users can get information of interest by typing “HLA Allele”, “Aligned Allele”, “Gene Pair” or upload a file as the example in 3DHLAMatch page. (ii) Sequence Alignment This module aims to help users get the information on HLA sequence differences. Users can use the ClustalW2 tool to achieve sequence alignment between mismatched HLA donors and receivers. (iii) 3D-View This module aims to provide users with the function of visualization of HLA sequence mismatch sites in 3D structure. Users can upload PDB files of HLA to 3Dmol, and visualize sequence differences of HLA in three-dimensional structure by setting “Chain” and “Residue”. (iv) Risk report This module aims to provide users with the assessment of transplant risks according to the function of mismatch sites. The Risk Site page in HLA3D provides information on HLA mismatch sites associated with acute graft rejection host disease (aGvHD) after transplantation, as well as the annotated information of the sites recognized by peptide and T cell receptor (TCR) in HLA molecule. Users can enter “Risk Position” and “Alignment Position” on the Risk Site page to query the risk information of mismatched sites. “Risk Position” refers to sites with structural differences of HLA molecules, while “Alignment Position” refers to sites with sequence differences of HLA molecules.
Introduction
Tumor neoantigens are a hot topic in the field of tumor immunotherapy, but not all tumor mutations can produce immunogenic antigenic peptides. Here, we took into account the key characteristics that affect the immunogenicity of the peptide and designed the Antigenic Peptide Prediction system by incorporating open source and our own software.
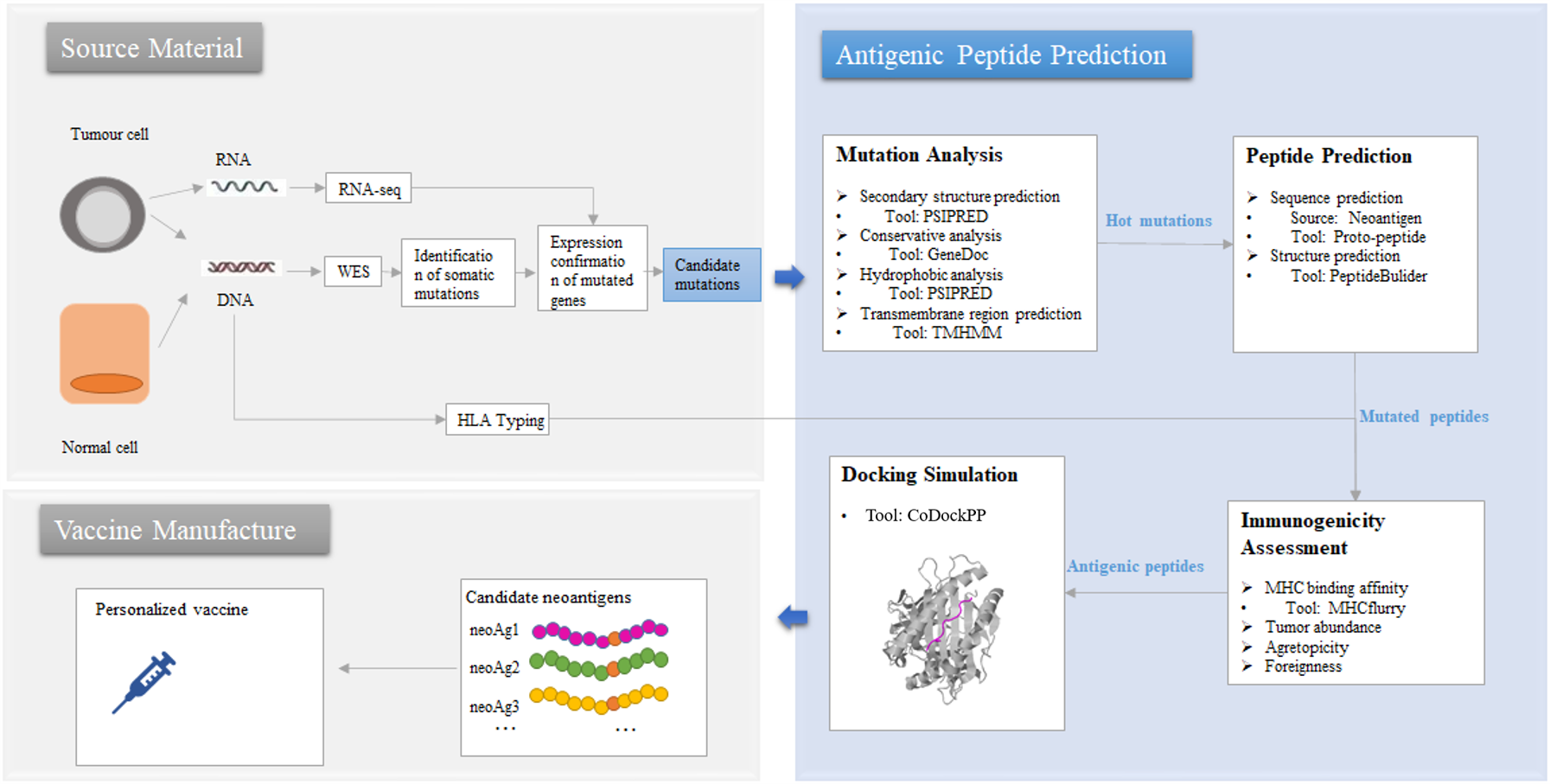
Figure 2. The prediction process of Antigenic Peptide Prediction pipeline in HLA3D
As the picture shows, Antigenic Peptide Prediction Pipeline provides users with the following functions: (i) Mutation Analysis This module aims to help users narrow the range of candidate mutations. Mutation page in HLA3D provides users with three types of mutation data associated with tumorigenesis. Users can browse the sub-pages of the Mutation page: SMG, HLA and HBV to obtain the hot spot mutations of the significant mutation genes (SMG), HLA and HBV virus related to different tumor respectively, and can also use the PSRPRED tool in HLA3D to predict the secondary structure and find the hot spot mutations in key regions of the gene. We have previously used tools such as GeneDoc and TMHMM to analyze the conserved and transmembrane regions of the sequences, and users can also use other tools to achieve more comprehensive predictive analysis. (ii) Peptide Prediction This module aims to help users obtain the sequence and structure of the mutated peptide. Peptide Sequence Prediction Users can browse the Neoantigen page, which stores more than 340,000 potential neoantigens for 11 common tumors that we have predicted based on the affinity between 989 hot spot mutations and all common HLA class Ⅰ genes in HLA3D. We also provide users with the Proto-Peptide tool, which they can use by entering "Protein Sequence" and setting parameters of "Original Residue," "Position," "New Residue," and "Length“ to obtain overlapping peptides containing mutations, which can be used as input to tools such as NetMHCpan and MHCflurry for scanning and prediction of HLA epitopes. Peptide Structure Prediction According to the functions of PeptideBulider package, we design a convenient input interface for users. PeptideBulider supports two ways to generate peptide structures. On the one hand, users can input a peptide sequence, generate an extended structure with default values for the backbone dihedral angles (ϕ = −120∘, ψ = 140∘, ω = 180∘). On the other hand, users can also custom Bond angles and bond lengths for every residue to create a specific conformation, and the first residue input should in geometry form. Notably, PeptideBulider do not provide any tools for energy minimization or rotamer packing. (iii) Immunogenicity Assessment This module aims to help users assess the potential immunogenicity of the mutated peptides and narrow the range of antigenic peptides. We designed a convenient input interface based on the key functions of MHCflurry to help users assess binding affinity, antigen processing, and presentation of the mutated peptides. Users can complete the prediction by using the method of “MHCfurry Predict” and “MHCfurry Predict Scan” in HLA3D. (iv) Docking Simulation This module aims help users get the initial conformation of peptide-HLA docking. Users can upload the peptide and HLA structure through CodockPP, and predict the initial docking conformation for subsequent dynamic simulation and so on. First, users can dock the peptide to the HLA in Ambiguous type without any site constraints. Second, multiple docking between peptide and HLA molecule can also be performed by inputting several sites (total sites < 8) on HLA molecule and peptide.
Introduction
ClustalW2 is a multi-sequence alignment program for DNA or protein. Clustal is a progressive alignment method. A distance matrix is constructed by pairwise alignment of multiple sequences, and then the phylogenetic tree is generated to weight the closely related sequences. Alignment starts with the two most closely related sequences, gradually introducing adjacent sequences and continually reconstructing the comparison until all sequences have been added. ClustalW2 in HLA3D allows users to compare sequences of different HLA genotypes to find amino acid differences.
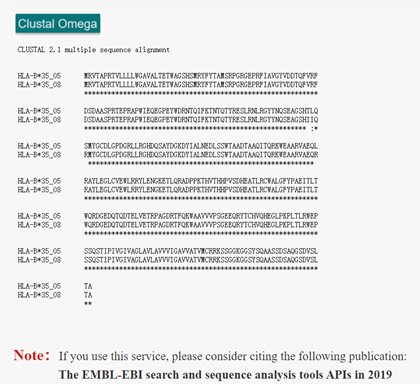
How to use this tool? Step-1 Enter Query Sequence Users should enter query sequence in FASTA format directly into the input data box or upload a sequence file in FASTA format. i. FASTA format The first character on the first line of a FASTA file is ">", followed by any text description, for sequence marking. The sequence itself begins on the second line. In general, nucleotides are written in both upper and lower case, while amino acids are written in capital letters. Step-2 Submit After clicking Submit, the page will display the sequence of comparison and the result of comparison. The asterisk (*) indicates that the sequence is consistent, the colon (:) indicates that the sequence is of high similarity, and the dot (.) indicates that the sequence similarity is low.
For example Input: >HLA-B*35:05 MRVTAPRTVLLLLWGAVALTETWAGSHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRAPWIEQEGPEYWDRNTQIFKTNTQTYRESLRNLRGYYNQSEAGSHTLQSMYGCDLGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQRADPPKTHVTHHPVSDHEATLRCWALGFYPAEITLTWQRDGEDQTQDTELVETRPAGDRTFQKWAAVVVPSGEEQRYTCHVQHEGLPKPLTLRWEPSSQSTIPIVGIVAGLAVLAVVVIGAVVATVMCRRKSSGGKGGSYSQAASSDSAQGSDVSLTA >HLA-B*35:08 MRVTAPRTVLLLLWGAVALTETWAGSHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRTEPRAPWIEQEGPEYWDRNTQIFKTNTQTYRESLRNLRGYYNQSEAGSHIIQRMYGCDLGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQRRAYLEGLCVEWLRRYLENGKETLQRADPPKTHVTHHPVSDHEATLRCWALGFYPAEITLTWQRDGEDQTQDTELVETRPAGDRTFQKWAAVVVPSGEEQRYTCHVQHEGLPKPLTLRWEPSSQSTIPIVGIVAGLAVLAVVVIGAVVATVMCRRKSSGGKGGSYSQAASSDSAQGSDVSLTAOutput:Figure 1. The ClustalW2 alignment result of HLA-B*35:08 and HLA-B*35:05 in HLA3D.
Reference
1. Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019 Jul 2;47(W1):W636-W641. doi: 10.1093/nar/gkz268.
2. Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7:539 doi:10.1038/msb.2011.75
3. Sievers F, Higgins DG (2018) Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27:135-145
4. Sievers F, Barton GJ, Higgins DG (2020) Multiple Sequence Alignment. Bioinformatics 227, pp 227-250, AD Baxevanis, GD Bader, DS Wishart (Eds)
Introduction
3dmol.js is a JavaScript library for visualizing the three-dimensional structure of molecules. 3dmol.js provides an API for developers to make it easy for users to share and embed molecular data on websites. Here, we designed a convenient input interface based on the function of 3Dmol to help the user locate amino acid residues in the three-dimensional structure.
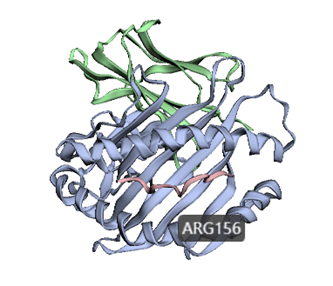
How to use this tool? Step-1 Upload HLA structure file Users can upload PDB files of HLA proteins. Step-2 Set parameters The user needs to set the parameters of amino acid residues to realize the visualization of the site in the three-dimensional structure. i. Chain Enter the name of the protein chain, such as "A". ii. Residue Enter the position of the amino acid residues to be visualized, separated by commas, such as “19,23,26”. Step-3 Submit Click Submit and the page will return the predicted results. The 3Dmol visualizations are sometimes slow to load and are recommended to be opened in a Google browser.
For example Input:Output:Figure 3. The visualization result of ARG156 residue in chain A of HLA-B*35:05 structure (PDB ID: 4PRB) by 3Dmol in HLA3D.

Reference:
Nicholas Rego and David Koes
3Dmol.js: molecular visualization with WebGL
Bioinformatics (2015) 31 (8): 1322-1324 doi:10.1093/bioinformatics/btu829
Introduction
Based on the position-specific score matrix generated by PSIBLAST, PSIPRED uses a two-layer neural network to predict the secondary structure of proteins. PSIPRED in the HLA3D Toolkit helps users predict the secondary structure of any sequence and find the mutation sites of interest.
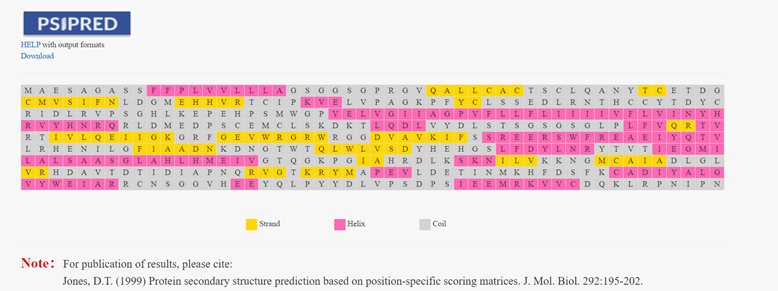
How to use this tool? Step-1 Enter Query Sequence Users should enter query sequence in FASTA format directly into the input data box or upload a sequence file in FASTA format. Step-2 Enter Email (optional) The user can enter an email address, and when the PSIPED job finishes, the user will be sent an email with the Job ID and other information Step-3 Submit After clicking Submit, the page will return the prediction result of the secondary structure of this sequence. The secondary structure of the protein, such as helix, coil, and stand, is highlighted in different colors. Step-4 Check Result Sometimes the PSRPRED Job takes a long time to run, and the user can query the predicted results by entering the Job ID in the email after the Job has finished running.
For example Input sequence: >sp|P36896|ACV1B_HUMAN Activin receptor type-1B OS=Homo sapiens OX=9606 GN=ACVR1B PE=1 SV=1 MAESAGASSFFPLVVLLLAGSGGSGPRGVQALLCACTSCLQANYTCETDGACMVSIFNLD GMEHHVRTCIPKVELVPAGKPFYCLSSEDLRNTHCCYTDYCNRIDLRVPSGHLKEPEHPS MWGPVELVGIIAGPVFLLFLIIIIVFLVINYHQRVYHNRQRLDMEDPSCEMCLSKDKTLQ DLVYDLSTSGSGSGLPLFVQRTVARTIVLQEIIGKGRFGEVWRGRWRGGDVAVKIFSSRE ERSWFREAEIYQTVMLRHENILGFIAADNKDNGTWTQLWLVSDYHEHGSLFDYLNRYTVT IEGMIKLALSAASGLAHLHMEIVGTQGKPGIAHRDLKSKNILVKKNGMCAIADLGLAVRH DAVTDTIDIAPNQRVGTKRYMAPEVLDETINMKHFDSFKCADIYALGLVYWEIARRCNSG GVHEEYQLPYYDLVPSDPSIEEMRKVVCDQKLRPNIPNWWQSYEALRVMGKMMRECWYAN GAARLTALRIKKTLSQLSVQEDVKI Output:Figure 2. The prediction result of PSRPRED in HLA3D.
Reference:
1. Jones, D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195-202.
Introduction
Proto-Peptide, written in the Java language, is a sequence processing tool designed to help users obtain overlapping peptides that contain mutations.
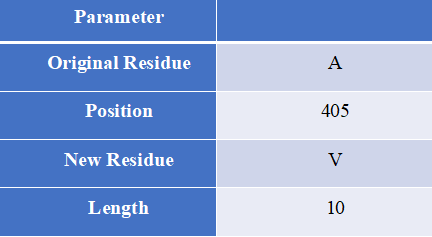
How to use this tool? Step-1 Enter Query Sequence Users should enter predict sequence in FASTA format directly into the input data box or upload a sequence file in FASTA format. Step-2 Set parameters Users can set different parameters to generate custom peptides containing mutations for HLA epitope scanning. Input explanation i. Original Residue Amino acids before mutation ii. Position Location of mutation iii. New Residue Mutated amino acids iv. Length The length of the peptide containing the mutation Step-3 Submit After clicking Submit, the page will return an overlapping peptide of a certain length containing a mutation and a wild peptide of the same length. Output Explanation The peptide sequence is provided to the user in FASTA format. “>WT” labeled wild peptides, “>Mut” marked the mutant peptide, followed by the mutation information of the sequence, which consists of the gene name and mutation.
For example Input sequence: >sp|P36896|ACV1B_HUMAN Activin receptor type-1B OS=Homo sapiens OX=9606 GN=ACVR1B PE=1 SV=1 MAESAGASSFFPLVVLLLAGSGGSGPRGVQALLCACTSCLQANYTCETDGACMVSIFNLD GMEHHVRTCIPKVELVPAGKPFYCLSSEDLRNTHCCYTDYCNRIDLRVPSGHLKEPEHPS MWGPVELVGIIAGPVFLLFLIIIIVFLVINYHQRVYHNRQRLDMEDPSCEMCLSKDKTLQ DLVYDLSTSGSGSGLPLFVQRTVARTIVLQEIIGKGRFGEVWRGRWRGGDVAVKIFSSRE ERSWFREAEIYQTVMLRHENILGFIAADNKDNGTWTQLWLVSDYHEHGSLFDYLNRYTVT IEGMIKLALSAASGLAHLHMEIVGTQGKPGIAHRDLKSKNILVKKNGMCAIADLGLAVRH DAVTDTIDIAPNQRVGTKRYMAPEVLDETINMKHFDSFKCADIYALGLVYWEIARRCNSG GVHEEYQLPYYDLVPSDPSIEEMRKVVCDQKLRPNIPNWWQSYEALRVMGKMMRECWYAN GAARLTALRIKKTLSQLSVQEDVKI Set parameters:Output:Figure 4. The prediction result of Proto-Peptide tool in HLA3D.

Introduction
MHCFlurry is a predictive model for the binding affinity of peptides to MHC class Ⅰ-like alleles, covering approximately 14,000 MHCI alleles in humans and a handful of other species. In addition, MHCFlurry has introduced two other predictors, an "antigen processing" predictor that attempts to simulate MHC allele-independent effects, such as proteosome cleavage, and a "presentation" predictor that combines processing predictions with binding affinity predictions to give a composite "presentation score." We designed a user-friendly input interface based on the key features of MHCFlurry. There are two types of predictions that can be implemented: "MHCFlurry Predict" and "MHCFlurry Predict Scan".
How to use this tool? (1)If you want to predict the binding affinity of individual peptides to MHC molecules, you can select "MHCFlurry predict" method to generate predictions. Step-1 Enter prediction information Users could enter prediction information directly into the input data box or upload a file in CSV, which should contain the “HLA allele”, “Peptide”, and, optionally, “n_flank” and “c_flank”. If you want to predict different peptides to MHC molecules, you can click the "+" button and a new line will appear on the input page. Step-2 Submit Click Submit and the page will return the predicted results along with a download link. Input explanation i. allele You can put in an HLA Allele and make a single gene prediction. And you can also give a comma separated list of HLA alleles. In this case, the tightest binding affinity across the alleles for the sample will be returned. ii. peptide Enter the predicted peptide, one peptide in a row. iii. n_flank The upstream and downstream sequences of the peptides from their source proteins. If you want to generate more accurate cleavage prediction, you could input the n_flank information of the peptide. iv. c_flank The downstream sequences of the peptides from their source proteins If you want to generate more accurate cleavage prediction, you could input the c_flank information of the peptide. Output Explanation i. mhcflurry_affinity The binding affinity predictions are given as affinities (KD) in nM in the mhcflurry_affinity column. Lower values indicate stronger binders. A commonly-used threshold for peptides with a reasonable chance of being immunogenic is 500 nM. ii. mhcflurry_affinity_percentile The mhcflurry_affinity_percentile gives the percentile of the affinity prediction among a large number of random peptides tested on that allele (range 0 - 100). Lower is stronger. Two percent is a commonly-used threshold. iii. mhcflurry_processing_score These range from 0 to 1 with higher values indicating more favorable processing or presentation. iv. mhcflurry_presentation_score These range from 0 to 1 with higher values indicating more favorable processing or presentation. v. mhcflurry_presentation_percentile The mhcflurry_presentation_percentile gives the percentile of the presentation prediction among a large number of random peptides tested on that allele (range 0 - 100). Lower is stronger. (2)If you want to scan protein sequences for epitopes, you can select "MHCFlurry predict scan" method to generate predictions. Step-1 Enter prediction information Input explanation i. allele Users can put in an HLA Allele and make a single gene prediction. And you can also give a comma separated list of HLA alleles. In this case, the tightest binding affinity across the alleles for the sample will be returned. ii. peptide Enter the predicted protein sequence in FSATA format or upload a file in FASTA format. Step-2 Submit Click Submit and the page will return the predicted results along with a download link.
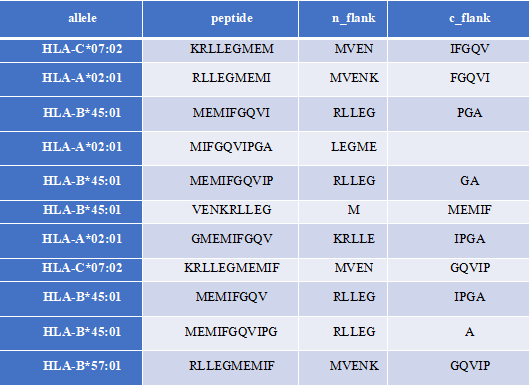
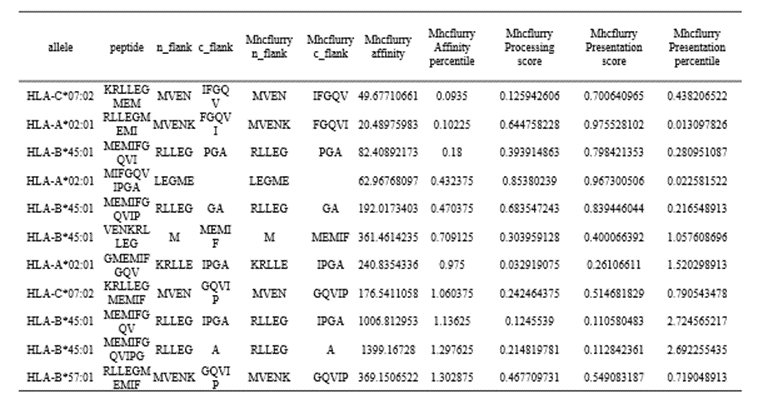
For example1For example2Input:
Output:
Figure 5. The prediction result of “MHCflurry predict” method in HLA3D
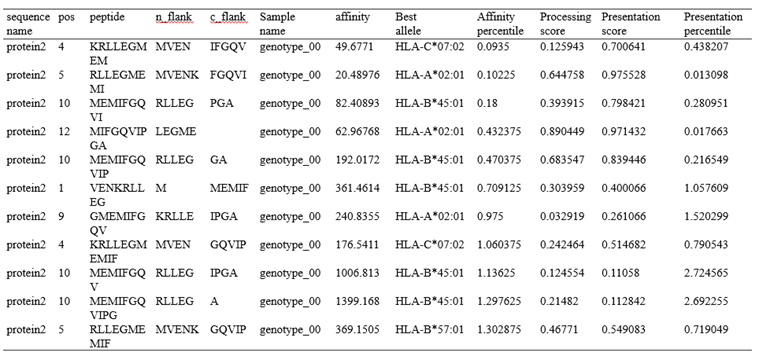
Input:
HLA allele: HLA-A*02:01,HLA-A*03:01,HLA-B*57:01,HLA-B*45:01,HLA-C*02:01,HLA-C*07:02 Protein sequence in FASTA format: >protein1 MSSSSTPVCPNGPGNCQV >protein2 MVENKRLLEGMEMIFGQVIPGAOutput:
Figure 6. The prediction result of “MHCflurry predict scan” method in HLA3D.
Reference:
1. T. J. O’Donnell, et al. “MHCflurry 2.0: Improved pan-allele prediction of MHC I-presented peptides by incorporating antigen processing,” Cell Systems, 2020. https://doi.org/10.1016/j.cels.2020.06.010
2. T. J. O’Donnell, et al., “MHCflurry: Open-Source Class I MHC Binding Affinity Prediction,” Cell Systems, 2018. https://doi.org/10.1016/j.cels.2018.05.014
Introduction
PeptideBuilder is a Python package that can be used to build any peptide conformation. One can add a residue to the C-terminal of an existing polypeptide model or generate a conformation of a single amino acid. Considering that peptides presented by different MHC molecules present different conformations, we designed and provided users with two conformational peptides schemes according to its function.
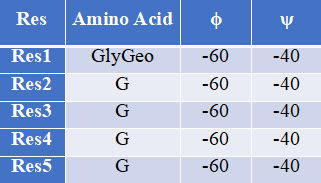
How to use this tool? (1)If you want to generate an extended peptide conformation, you can directly entry a peptide sequence and use the default backbone dihedral Angle (ϕ= −120∘, ψ = 140∘, ω = 180∘). The default values for bond lengths and angles were obtained by measuring these quantities in a large collection of published crystal structures and recording the average for each quantity. Step-1 Enter peptide sequence Fill in the input box with the sequence of the peptide, for example, “TLACFVLAAV”. Step-2 Add terminal oxygen If you want to add terminal oxygen (OXT) to the final residue, you could check this option. Step-3 Submit Click Submit and the page will return the predicted results along with a download link. (2)If you want to generate a specific peptide conformation, you can also customize bond angles and bond lengths for each residue in the peptide chain to create a specific conformation. Step-1 Start with the first amino acid residue The formation of a peptide with a specific conformation begins with the first amino acid residue of the peptide. To enter the geometric conformation of the amino acid, the user can pull down the "Please Select" button to select the amino acid residues, such as “ThrGeo”. Then, you can input the values of backbone dihedral Angle, such as, ϕ= “−120”, ψ = “140”. Step-2 Add amino acid residues Click the "+" button on the page, and an additional line will appear in the Input box. You can enter amino acid residues, such as “A”, and the backbone dihedral Angle, such as, ϕ= “−120”, ψ = “140”. Step-3 Add terminal oxygen If you want to add terminal oxygen (OXT) to the final residue, you could check this option. Step-4 Submit Click Submit and the page will return the predicted results along with a download link.
For example1 Input peptide sequence: TLACFVLAAV Output: The extended peptide conformation generated by PeptideBuilder.For example2 Input:Figure 7. The peptide (sequence: TLACFVLAAV) conformation shown by PyMol(Schrödinger Software Release 2021-2)
Figure 8. The specific peptide conformation of an alpha helix consisting of five glycines, shown by PyMol (Schrödinger Software Release 2021-2).
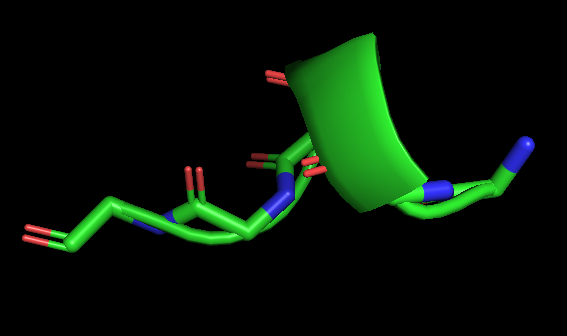
Reference:
1. Tien MZ, Sydykova DK, Meyer AG, Wilke CO. PeptideBuilder: A simple Python library to generate model peptides. PeerJ. 2013 May 21;1:e80. doi: 10.7717/peerj.80.
2. Tien MZ, Meyer AG, Sydykova DK, Spielman SJ, Wilke CO. Maximum allowed solvent accessibilites of residues in proteins. PLoS One. 2013 Nov 21;8(11):e80635. doi: 10.1371/journal.pone.0080635.
Introduction
CoDockPP is a tool that can be used for protein-protein docking. The program adopts knowledge-based scoring function to evaluate the docking posture to ensure the accuracy of docking results. In addition, the program allows the user to set the site constraint information on the recipient and donor proteins respectively, improving the efficiency and success rate of docking. Considering that the binding between the peptide and HLA molecules mainly depends on the interaction between the main anchor residues of the peptide and the conserved hydrogen bond network in the B and F pockets of the HLA antigen-binding groove, we modified the input of CoDockPP software to provides users with more realistically simulate schemes for the binding of peptides and HLA molecules.
How to use this tool? Step-1 Input HLA protein Users need to upload the input coordinates of the HLA protein (large) in strict PDB format. Before uploading a PDB file, you should use a PDB checker (for example, Molprobity) to anticipate and fix any potential PDB errors. Step-2 Input peptide Users need to upload the input coordinates of the peptide (large) in strict PDB format. Similarly, a PDB inspector (for example, Molprobity) should be used to predict and fix any potential PDB errors before uploading a PDB file. Step-3 Enter your email You can enter an E-mail address to receive a link to access docked results later. Step-4 Set site constraint CodockPP software can perform global docking and site-specific docking to predict the binding complexes between two proteins. You can enter one constraint residue on the receptor interface and another one on the ligand interface. Constraint conditions can be set on both HLA protein and peptide structures. The total number of sites is <8. i. Site Such as "Leu A 77", which represents the residue LEU77 of the chain A on the protein. Please use commas (,) to separate different sites. ii. Constraint Type The site constraints can be set as ambiguous constraint and multiple constraint. When you choose ambiguous constraint, the conformation is required with at least one site on the interface of receptor or ligand. When you choose multiple constraint, the conformation is retained with both of the two sites on the interface. Step-4 Submit Click Submit and the page will return the predicted results along with a download link. Step-5 Check Result Query the docking Result at “Check Result” with the “Job ID”. Note: More tutorials and explanations are available on CoDockPP's website: http://codockpp.schanglab.org.cn
Reference:
1. Kong R, Wang F, Zhang J, Xu X J, Chang S. CoDockPP: a multistage approach for global and site-specific protein-protein docking. Journal of Chemical Information and Modeling, 2019, 59(8): 3556-3564.
2. Kong R, Liu R R, Xu X M, Zhang D W, Xu X S, Shi H, Chang S. Template‐based modeling and ab‐initio docking using CoDock in CAPRI. Proteins-Structure Function And Bioinformatics, 2020, 88(8): 1100-1109.
3. Lensink M F, Brysbaert G, Nadzirin N, Velankar S, Chaleil R A G, Gerguri T, Bates P A, Laine E, Carbone A, Grudinin S, Kong R, Liu R R, Xu X M, Shi H, Chang S, et al. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins-Structure Function And Bioinformatics, 2019, 87(12): 1200-1221.
Submission
You are welcome to submit HLA structures to HLA3D to facilitate data sharing!References
[1] Robinson, J., Barker, D. J., Georgiou, X., et al., IPD-IMGT/HLA Database. Nucleic Acids Res 2020, 48 (D1), D948-D955.[2] Burley, S. K., Bhikadiya, C., Bi, C., et al., RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 2021, 49 (D1), D437-D451.
[3] Gonzalez-Galarza, F. F., McCabe, A., Santos, E., et al., Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res 2020, 48 (D1), D783-D788.
[4] Wang, J., Huang, D., Zhou, Y., et al., CAUSALdb: a database for disease/trait causal variants identified using summary statistics of genome-wide association studies. Nucleic Acids Res 2020, 48 (D1), D807-D816.
[5] Shukla, S. A., Rooney, M. S., Rajasagi, M., et al., Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 2015, 33 (11), 1152-8.
[6] Kandoth, C., McLellan, M. D., Vandin, F., et al., Mutational landscape and significance across 12 major cancer types. Nature 2013, 502 (7471), 333-339.
[7] Rego, N.; Koes, D., 3Dmol.js: molecular visualization with WebGL. Bioinformatics 2015, 31 (8), 1322-4.
[8] Jones, D. T., Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999, 292 (2), 195-202.
[9] Madeira, F., Park, Y. m., Lee, J., et al., The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research 2019, 47 (W1), W636-W641.
[10] Tien, M. Z., Sydykova, D. K., Meyer, A. G., et al., PeptideBuilder: A simple Python library to generate model peptides. PeerJ 2013, 1, e80.
[11] Kong, R., Wang, F., Zhang, J., et al., CoDockPP: A Multistage Approach for Global and Site-Specific Protein-Protein Docking. J Chem Inf Model 2019, 59 (8), 3556-3564.
[12] Lensink, M. F.; Brysbaert, G.; Nadzirin, N., et al., Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 2019, 87 (12), 1200-1221.
[13] Kong, R., Liu, R. R., Xu, X. M., et al., Template-based modeling and ab-initio docking using CoDock in CAPRI. Proteins 2020, 88 (8), 1100-1109.
[14] O'Donnell, T. J., Rubinsteyn, A., Bonsack, M., et al., MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst 2018, 7 (1), 129-132 e4.
[15] O'Donnell, T. J.; Rubinsteyn, A.; Laserson, U., MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst 2020, 11 (1), 42-48 e7.
[16] Gu, S., Lv, L., Lin, X., et al., Using structural analysis to explore the role of hepatitis B virus mutations in immune escape from liver cancer in Chinese, European and American populations. J Biomol Struct Dyn 2020, 1-11.
[17] Jurtz, V., Paul, S., Andreatta, M., et al., NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 2017, 199 (9), 3360-3368.
[18] Wells, D. K., van Buuren, M. M., Dang, K. K., et al., Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183 (3), 818-834 e13.
[19] Heemskerk, M. B., Roelen, D. L., Dankers, M. K., et al., Allogeneic MHC class I molecules with numerous sequence differences do not elicit a CTL response. Hum Immunol 2005, 66 (9), 969-76.
[20] Mack, S. J., Cano, P., Hollenbach, J. A., et al., Common and well-documented HLA alleles: 2012 update to the CWD catalogue. Tissue Antigens 2013, 81 (4), 194-203.
[21] Sanchez-Mazas, A., Nunes, J. M., Middleton, D., et al., Common and well-documented HLA alleles over all of Europe and within European sub-regions: A catalogue from the European Federation for Immunogenetics. HLA 2017, 89 (2), 104-113.
[22] He, Y., Li, J., Mao, W., et al., HLA common and well-documented alleles in China. HLA 2018, 92 (4), 199-205.